We can get the DNA sequence of the range of chromosome in human genome by using ucsc web tool.
http://genome.ucsc.edu/cgi-bin/das/hg19/dna?segment=chr8:126544471,126546471


We can get the DNA sequence of the range of chromosome in human genome by using ucsc web tool.
http://genome.ucsc.edu/cgi-bin/das/hg19/dna?segment=chr8:126544471,126546471
We can simply use the following formulae to extract the number from string in Excel file

=SUMPRODUCT(MID(0&E4, LARGE(INDEX(ISNUMBER(--MID(E4, ROW(INDIRECT("1:"&LEN(E4))), 1)) * ROW(INDIRECT("1:"&LEN(E4))), 0), ROW(INDIRECT("1:"&LEN(E4))))+1, 1) * 10^ROW(INDIRECT("1:"&LEN(E4)))/10)
Note: Tested in Excel 2010
We can use the tool picard to liftOver VCF file by batch
URL:
http://broadinstitute.github.io/picard/command-line-overview.html#LiftoverVcf

create dictionary (optional)
if you do not have the dict of reference fasta, you need to create first
java -jar /software/picard-tools-2.8.1/picard.jar CreateSequenceDictionary REFERENCE=/software/GATK_files/hg38/hg38.fa OUTPUT=/software/GATK_files/hg38/hg38.dict
download the chain file
Go the http://hgdownload.cse.ucsc.edu/downloads.html#human and download from liftOver hyperlink
liftOver
java -jar /software/picard-tools-2.8.1/picard.jar LiftoverVcf I=input.vcf O=lifted_over.vcf CHAIN=/software/liftOver/hg19ToHg38.over.chain.gz REJECT=rejected_variants.vcf R=/software/GATK_files/hg38/hg38.fa
Reference:
Chain format
URL: https://genome.ucsc.edu/goldenpath/help/chain.html
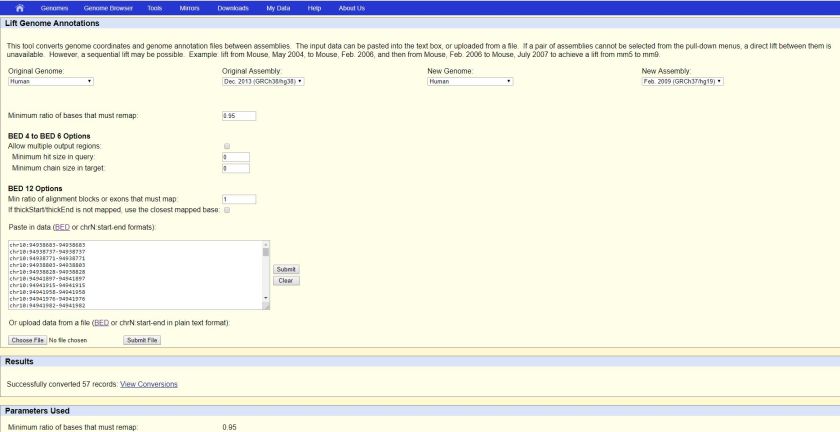
We can visit the web tool at https://genome.ucsc.edu/cgi-bin/hgLiftOver
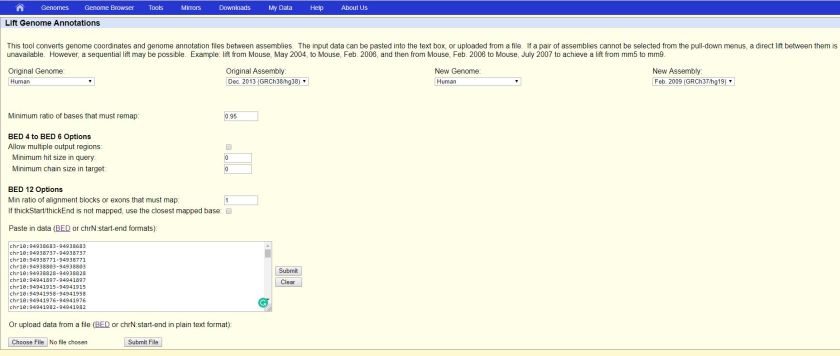
Select the Original Genome, Original Assembly, New Genome and New Assembly, say
Original Genome: Human
Original Assembly: Dec. 2013 (GRCh38/hg38)
New Genome: Human
New Assembly: Feb. 2009 (GRCh37/hg19)
Paste the data and click “Submit” button as the RHS of the text area
The result file (i.e. hglft_genome_260a_37bd80.bed) will be ready to download after some time
/software/samtools/1.8/bin/samtools bam2fq -N AGG0011.bam > AGG0011.fastq
cat AGG0011.fastq | grep ‘^@.*/1$’ -A 3 –no-group-separator > AGG0011_R1.fastq
cat AGG0011.fastq | grep ‘^@.*/2$’ -A 3 –no-group-separator > AGG0011_R2.fastq
I want to download the FASTQ files from Basespace to the Linux server directly without first downloading to local PC based on the project. I found three references:
1. Download files from Illumina’s BaseSpace
https://gist.github.com/lh3/54f535b11a9ee5d3be8e
2. Use the Python Run Downloader
https://help.basespace.illumina.com/articles/tutorials/using-the-python-run-downloader/
3. API
https://developer.basespace.illumina.com/docs/content/documentation/rest-api/api-reference
But, the Python Run Downloader will download files based on the Run Id that I want to download based on the project. Therefore, I modified the script to meet my requirement.
from urllib2 import Request, urlopen, URLError
import json
import math
import sys
import os
import socket
import optparse
def arg_parser():
cwd_dir = os.getcwd()
parser = optparse.OptionParser()
parser.add_option( '-p', dest='projid', help='Project ID: required')
parser.add_option( '-a', dest='accesstoken', help='Access Token: required')
( options, args ) = parser.parse_args()
try:
if options.projid == None:
raise Exception
if options.accesstoken == None:
raise Exception
except Exception:
print("Usage: BaseSpaceRunDownloader_vN.py -p <ProjID> -a <AccessToken>")
sys.exit()
return options
def restrequest(rawrequest):
request = Request(rawrequest)
try:
response = urlopen(request)
json_string = response.read()
#print(json_string)
json_obj = json.loads(json_string)
except URLError, e:
print 'Got an error code:', e
sys.exit()
return json_obj
def downloadrestrequest(rawrequest,path):
dirname = ProjID + os.sep + os.path.dirname(path)
#print(dirname)
if dirname != '':
if not os.path.isdir(dirname):
os.makedirs(dirname)
request = (rawrequest)
outfile = open(ProjID + os.sep + path,'wb')
try:
response = urlopen(request,timeout=1)
outfile.write(response.read())
outfile.close()
except URLError, e:
print 'Got an error code:', e
outfile.close()
downloadrestrequest(rawrequest,path)
except socket.error:
print 'Got a socket error: retrying'
outfile.close()
downloadrestrequest(rawrequest,path)
options = arg_parser()
ProjID = options.projid
AccessToken = options.accesstoken
hreflist = []
hrefcontentlist = []
pathlist = []
samplelist = []
#Step 1: Find the Biosample ID from project id first
#assume fewer than 1000 samples in a project
request = 'https://api.basespace.illumina.com/v2/biosamples?projectid=%s&access_token=%s&limit=1000' %(ProjID,AccessToken)
json_obj = restrequest(request)
nSamples = len(json_obj['Items'])
for sampleindex in range(nSamples):
sampleid = json_obj['Items'][sampleindex]['Id']
samplelist.append(sampleid)
samplecsv = ','.join([str(i) for i in samplelist])
print(samplecsv)
#Step 2: Call API to get datasets based on biosample
request = 'https://api.basespace.illumina.com/v2/datasets?inputbiosamples=%s&access_token=%s' %(samplecsv,AccessToken)
json_obj = restrequest(request)
totalCount = int(json_obj['Paging']['TotalCount'])
noffsets = int(math.ceil(float(totalCount)/1000.0))
for index in range(noffsets):
offset = 1000*index
request = 'https://api.basespace.illumina.com/v2/datasets?inputbiosamples=%s&access_token=%s&limit=1000&Offset=%s' %(samplecsv,AccessToken,offset)
#print(request)
json_obj = restrequest(request)
nDatasets = len(json_obj['Items'])
for fileindex in range(nDatasets):
href = json_obj['Items'][fileindex]['HrefFiles']
hreflist.append(href)
#Step 3: Get the download filepath (HrefContent) and filename (Path)
#normally two files per dataset in our case
for index in range(len(hreflist)):
request = '%s?access_token=%s'%(hreflist[index],AccessToken)
#print(request)
json_obj = restrequest(request)
nfiles = len(json_obj['Items'])
for fileindex in range(nfiles):
hrefcontent = json_obj['Items'][fileindex]['HrefContent']
hrefcontentlist.append(hrefcontent)
path = json_obj['Items'][fileindex]['Path']
pathlist.append(path)
for index in range(len(hreflist)):
request = '%s?access_token=%s'%(hrefcontentlist[index],AccessToken)
print(request)
print 'downloading %s' %(pathlist[index])
downloadrestrequest(request, pathlist[index])
The script can be started by
python BaseSpaceRunDownloader_v2.py -p $ProjId -a $AccessToken
Notes:
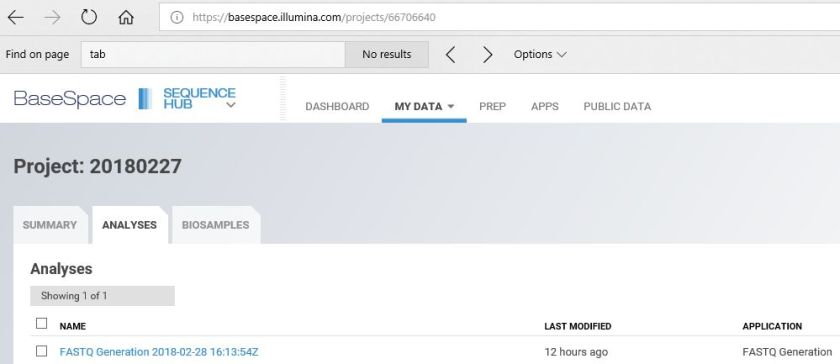
Files are saving to the subdirectory with directory name is <ProjId>
Hope it helps!!
In one of the project, we need to get the genomic sequence of a specified region. In this case, I use the API by ensemble
http://rest.ensembl.org/documentation/info/sequence_region
GET sequence/region/:species/:region
Returns the genomic sequence of the specified region of the given species. Supports feature masking and expand options.

In my case, I need to get the human (hg19), so I can simply paste the following URL on browser to get the result:
http://grch37.rest.ensembl.org/sequence/region/human/1:100000..100200:1?content-type=text/plain

If you have a list of range, you may write Excel Macro to help. For example, create a button and associate with the following VBA code.
After filling the chromosome and range in Colume A, B and C, and then click “Submit” button. The sequence will be shown in the Column D (Response)

Sub Button1_click()
Dim i As Integer
Dim Chr, StartPos, EndPos As String
Dim responseStr As String
'Skip header row
i = 2
Do While Cells(i, 1).Value <> ""
Chr = Cells(i, 1).Value
StartPos = Cells(i, 2).Value
EndPos = Cells(i, 3).Value
URL = "http://grch37.rest.ensembl.org/sequence/region/human/" & Chr & ":" & StartPos & ".." & EndPos & ":1?content-type=text/plain"
responseStr = http(URL)
Cells(i, 4).Value = responseStr
i = i + 1
Loop
MsgBox "Done!!"
End Sub
Function http(ByVal URL) As String
Dim MyRequest As Object
Set MyRequest = CreateObject("WinHttp.WinHttpRequest.5.1")
MyRequest.Open "GET", URL
' Send Request.
MyRequest.send
'And we get this response
http = MyRequest.responseText
End Function
Hope this helps!
I have a genomic range (chr9:129377235-132308843) and I would like to see which human genes are included in that range. Now, I am going to show how to do this by using the online tool TableBrowser (https://genome.ucsc.edu/cgi-bin/hgTables) in UCSC.

Click “get output” button to have the result:

You can load into Excel to manipulate the data.

Then, we can remove duplicates in Excel to get the distinct gene list.
Enjoy!!
I read many papers using PCA to show different clusters of the population but hard to see a step-by-step guide for a beginner like me. Here’re the steps I did.
I use mainly plink (version 1.9) and R (simple plot) on The Phase 2 HapMap as a PLINK fileset. The dataset can be download at http://zzz.bwh.harvard.edu/plink/res.shtml
Entire HapMap (release 23, 270 individuals, 3.96 million SNPs)
– CEU (release 23, 90 individuals, 3.96 million SNPs)
– YRI (release 23, 90 individuals, 3.88 million SNPs)
– JPT+CHB (release 23, 90 individuals, 3.99 million SNPs)
Since the dataset is already in plink format, I can simply run it against plink.
278673251 Sep 2 2008 hapmap_r23a.bed
114426472 Sep 2 2008 hapmap_r23a.bim
7470 Sep 2 2008 hapmap_r23a.fam
Step 1 :
You may get the following error when running the data directly by using plink
Error: .bim file has a split chromosome. Use –make-bed by itself to remedy this.
We can fix this by running the following:
plink --bfile hapmap_r23a --make-bed -out hapmap_r23a_sorted
Step 2 (QC):
You remove any individuals who have less than, say, 95% genotype data (–mind 0.05) and snps with a MAF<0.01 and a HWE P-value<0.00001 as well as a genotype failure rate greater than 0.1 are also removed.
plink --bfile hapmap_r23a_sorted --make-bed --mind 0.1 --maf 0.01 --geno 0.05 --hwe 0.00001 -out hapmap_r23a_cleaned
Step 3 (Prune):
First we prune the dataset to remove highly correlated variants, so that variants can be considered relatively independent, which is one of the assumptions we make in principal component analysis
plink --bfile hapmap_r23a_cleaned --indep-pairwise 50 5 0.2 --out LDprunedout
plink --bfile hapmap_r23a_cleaned --extract LDprunedout.prune.in --make-bed --out hapmap_r23a_ldpruned
Step 4 (PCA)
Execute the following script to do PCA
plink --bfile hapmap_r23a_ldpruned --pca --out pca_result
Files generated:
pca_result.hh
pca_result.eigenvec
pca_result.eigenval
pca_result.log
Step 5 (Plot)
This is straightforward in R.
1. Load the .eigenvec file. E.g. “eigenvec_table <- read.table(‘pca_result.eigenvec’)”
2. Use plot() on the columns you’re interested in. Top eigenvector will be in column 3 while second eigenvector will be in column 4, etc.. Therefore, if you want a set of pairwise plots covering the top 4 eigenvectors, you can use “plot(eigenvec_table[3:6])”.
>eigenvec_table <- read.table('pca_result.eigenvec') >plot(eigenvec_table[3:4])

This above plot should be fine and it is similar to the one plotted shown as follow by http://www.well.ox.ac.uk/gabriel-project using HapMap data, just no fancy graphics. Of course, you can plot beautifully by R but it is not target of this article.

Hope this helps!
I use bcftools version 1.6 where the latest version can be downloaded at https://samtools.github.io/bcftools/
Original file (test.genotypecalls.vcf) has 4 samples:

#!/bin/bash
file="test.genotypecalls.vcf"
for sample in `bcftools query -l $file`; do
bcftools view -c1 -Oz -s $sample -o ${file/.vcf*/.$sample.vcf.gz} $file
done
Results:
4 vcf.gz files have been generated.
test.genotypecalls.vcf
test.genotypecalls.A160251_Pro.vcf.gz
test.genotypecalls.A160252_Sib.vcf.gz
test.genotypecalls.A160253_Mot.vcf.gz
test.genotypecalls.A160254_Fat.vcf.gz
One sample [A160251_Pro] of split individual file content:

